
")
New Treatment for Spinal Muscular Atrophy
written by Dr Thomas Dunne
What is Spinal Muscular Atrophy?
Spinal Muscular Atrophy (SMA) is a rare genetic condition that causes progressive weakness and wasting of the muscles. It is a spectrum of conditions most commonly caused by a gene defect on chromosome 5q called the ‘survival motor neuron gene 1’, referred to as ‘SMN1’. With this gene being faulty, the individual is unable to produce enough of an important protein, called SMN, to have healthy nerves. This results in a reduction in signalling from the brain to the muscles, leading to a lack of movement which over time leads to wasting. It can affect muscles involved in movement, breathing, speaking and swallowing. Due to the existence of many types of SMA, each individual is affected differently, and will require different management and support. SMA does not affect intelligence or personality and is not a learning difficulty.
How does Spinal Muscular Atrophy Present?
SMA can present in a number of ways.
Common symptoms include:
- Movement difficulties e.g. difficulty walking, sitting-up, carrying objects
- Problems with breathing
- Problems with swallowing
Common signs include:
- Common signs include:
- Muscle weakness (flaccidity)
- Muscle wasting (atrophy)
- Low muscle tone (hypotonia)
- Muscle twitches (fasciculations)
- Loss of muscle reflexes (hyporeflexia)
What are the types of Spinal Muscular Atrophy?
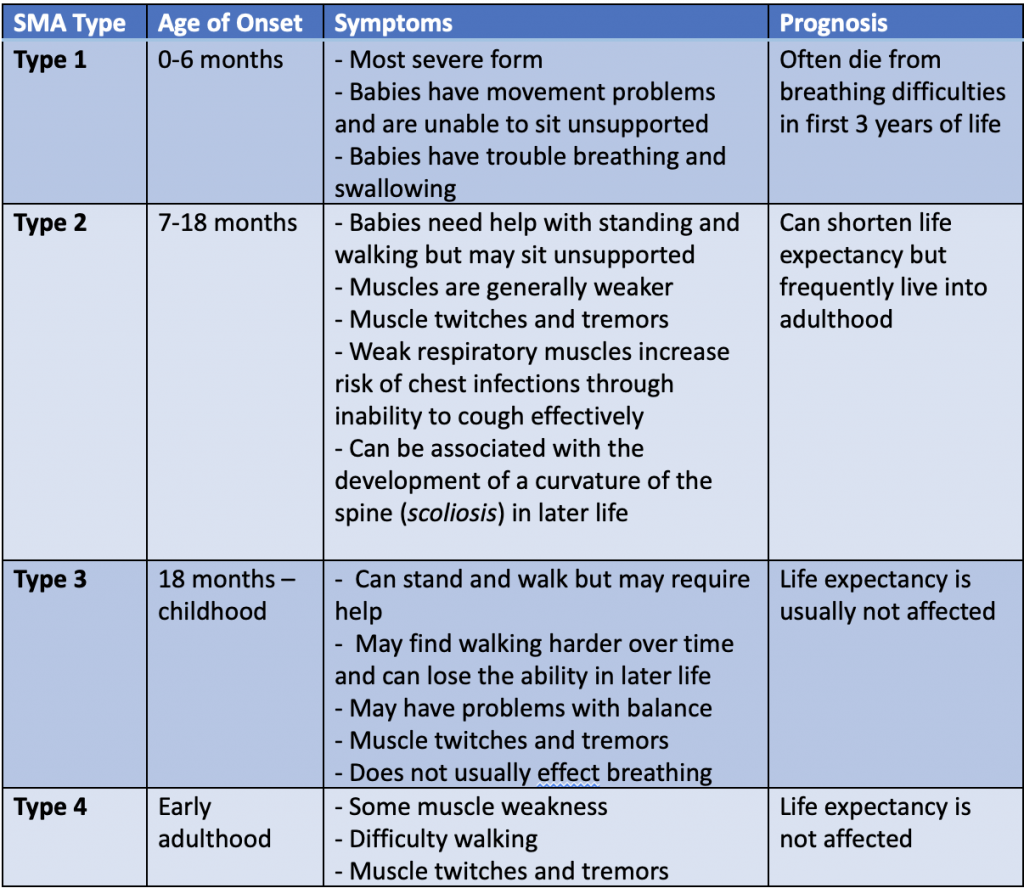
The main types of SMA are called types 1, 2, 3 and 4. These are all associated with a defective SMN1 gene. There are a number of other SMAs that are not associated with this gene. The below table outline the four main types of Spinal Muscular Atrophy:
There are some, even rarer, types of SMA that do not involve the SMN1 gene. A few examples of these are:
- X-linked spinal muscular atrophy. This is characterised by severe muscle weakness and breathing difficulty, and affected children often have misshapen bones.
- Spinal muscular atrophy with respiratory distress (SMARD). SMARD is usually diagnosed in the first year of life and causes severe respiratory problems.
- Spinobulbar muscular atrophy (SBMA) or Kennedy’s disease. SBMA predominantly affects men. It usually starts in middle age and does not affect life expectancy.
- Distal spinal muscular atrophy (DSMA). DSMA is a type of SMA that mainly affects the lower parts of the arms and legs, such as hands and feet.
More information on rarer types of SMA can be found on the SMA UK website.
How is Spinal Muscular Atrophy Diagnosed?
The most direct way to test for SMA is with a genetic blood test to confirm the faulty gene. As well as blood tests, there is sometimes the need for further investigations of nerve and muscle function. This can include:
- Specific blood tests for other neuromuscular conditions
- A muscle biopsy. This is where a needle is used to take a small sample of muscle tissue is removed for analysis.
- Electromyography (EMG). This is where small needs are directly inserted into a muscle to assess its function.
How is Spinal Muscular Atrophy Inherited?
SMA types 1-4 are autosomal recessive, which means that an individual must inherit two faulty genes (one from each parent) to have the condition. If a person has just one faulty gene, they are said to be a ‘carrier’. This is because that they themselves will not exhibit the symptoms of SMA, however if they have a baby with another carrier, their child is at risk of having SMA.
If two carriers of SMA have a child, there is a:
- 25% chance that their child will have SMA
- 25 % change that their child will not have SMA, and not be a carrier of SMA
- 50% chance that their child will not have SMA, but will be a carrier of SMA
For an individual that either has SMA themselves or has a family history of the disease, there can be a lot of questions when thinking about starting a family. Speaking to a genetic counsellor can help to explain the likelihood of their child having SMA. For those without SMA but with a family history of the disease, a blood test may help to determine whether they are a carrier. If you are pregnant and there is a risk of SMA, tests can be carried out on the embryo during pregnancy. The two techniques for this are called chorionic villous sampling (11-14 weeks pregnant) and amniocentesis (15-20 weeks pregnancy). Both tests are associated with a small increased risk of miscarriage. For those thinking of starting a family that have SMA, there are alternative options to traditional pregnancy. These include the use of donated sperm/eggs, or having their own eggs fertilised in a laboratory and testing the embryos for the faulty gene prior to implanting into the womb.
The SMA UK website provides a more detailed explanation of the genetic inheritance of SMA.
What are the treatment options for Spinal Muscular Atrophy?
Traditional therapies for SMA did not directly manage the condition and focused on improving quality of life. These remain vital today and provide huge benefit to people with SMA. These include:
- Exercises, to strengthen muscles and aid movement and breathing. A physiotherapist can become involved at this point.
- Assistive equipment, including mobility equipment within the home and walking aids e.g. leg braces. An occupational therapist can become involved at this point.
- Feeding help, including special diets and the use of a feeding tube. A dietician can become involved at this point.
- Breathing assistance, in the form of breathing masks and suction devices.
Recent advances have seen the introduction of the first medicine to directly target the cause of SMA. In June 2017, Nusinersen (SpinrazaTM) became Europe’s first approved disease-modifying medication for the treatment of SMA. It works through the SMN2 gene to produce more SMN protein. It is currently approved for use in SMA types 1, 2, 3, and 4. Clinical trials have been run in those with SMA types 1-3, but not yet in adults with SMA type 4. A second medication called ZolgensmaTM, is also being considered by the European Medicines Agency for licensing in Europe. It increases SMN protein production through the use of genetically engineered viruses. It is approved in the USA for use in children under 2-years that have a significant genetic risk of developing SMA. It is relatively early days in the pharmaceutical management of SMA, but recent developments are exciting and hopeful.
References and Useful Resources
- Spinal Muscular Atrophy UK. https://smauk.org.uk/.
- Spinal Muscular Atrophy, National Health Service. https://www.nhs.uk/conditions/spinal-muscular-atrophy-sma/
- Spinal Muscular Atrophy, Patient Info. https://patient.info/doctor/spinal-muscular-atrophy-pro#nav-1.
- Mercuri E, Finkel RS, Muntoni F et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018;28(2): 103-115.
- Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord 2018;28(3): 197-207.
- Guide to the 2017 International Standards of Care for SMA, ‘Spinal Muscular Atrophy UK’, ‘Cure SMA’, ‘SMA Europe’, ‘Muscular Dystrophy UK’ and ‘TREAT-NMD’. https://treat-nmd.org/wp-content/uploads/2019/06/uncategorized-A-Guide-to-the-2017-International-Standards-of-Care-for-SMA_UKEnglish_Digital-v2L.pdf.


14th August 2020 @ 9:21 am
My son Jackson, has type 2 spinal muscular atrophy. I cry everyday. I wish I could swap around our circumstances. I just pray he can be as happy as he can.
22nd April 2021 @ 8:34 pm
Dear Robert, I am so sorry to hear this and I hope you’re getting some peer-support so you can talk to those that find themselves in this same devastating situation. If you need suggestions of where to go for help please don’t hesitate to contact us on info@m4rd.org
Best wishes
Lucy
7th December 2020 @ 11:49 pm
After my MS diagnosis, my primary care provider introduced me to RichHerbs Foundation and their MS Formula protocol, the herbal treatment has made a tremendous difference for me. My symptoms including numbness and muscle weakness all disappeared after the treatment plan! Their website is w w w. richherbsfoundation. c o m..